Yesterday, October 1, marked the first day of implementation for PDUFA VI, the next iteration of the Prescription Drug User Fee Act (PDUFA) and one of the best tools that the U.S. Food and Drug Administration (FDA) has for enhancing regulatory efficiency and encouraging innovative development of safe and effective new medicines for patients.
First enacted in 1992, PDUFA allows the FDA to collect user fees from biopharmaceutical companies to help support the agency’s process for review of new drugs and biologics. Since its inception, PDUFA has helped make FDA review of new medicines more consistent, predictable and efficient while strengthening FDA’s already high safety and efficacy standards and increasing patient access to state-of-the-art treatments.
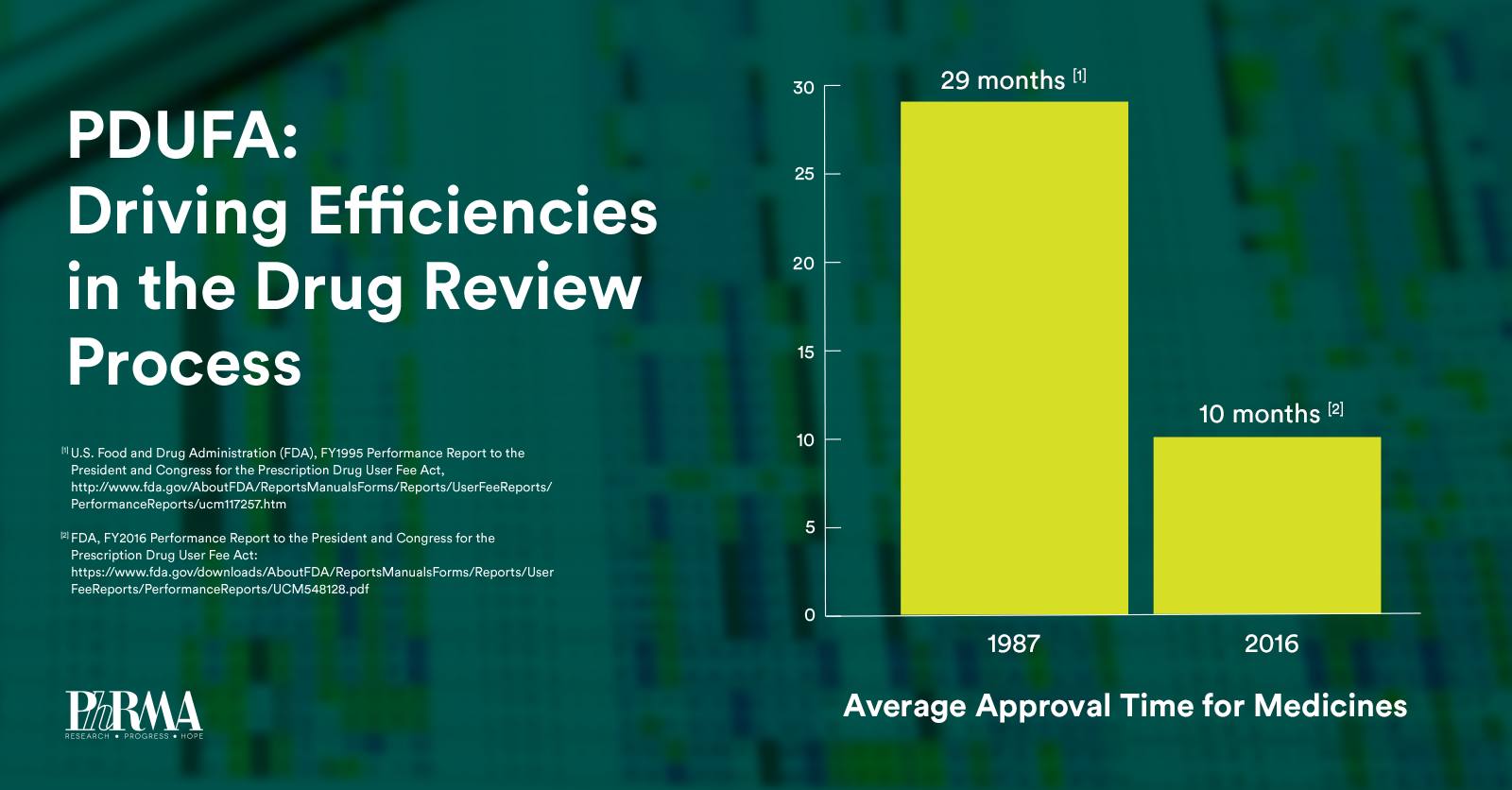
Over the next five years, PDUFA VI will build upon the regulatory science advances made in previous PDUFA cycles while providing sufficient resources to help maintain FDA’s “gold-standard” of medicine review. Specifically, PDUFA VI will:
- Enhance the predictability and efficiency of the drug review process;
- Support the development and application of 21st century regulatory approaches to drug development;
- Accelerate the integration of patient perspectives into the development and regulatory review of new medicines;
- Continue to strengthen post-approval safety; and
- Ensures that FDA can hire and retain a strong scientific and medical workforce to advance its public health mission.
Predictability and Efficiency of Drug Review Process
Each PDUFA reauthorization has strengthened the FDA’s drug review program with the goal of bringing safe and effective new medicines to patients in a timely manner. PDUFA VI builds on the success of the Breakthrough Therapy Program by investing dedicated resources to prioritize the development and availability of breakthrough medicines for patients with serious and life-threatening diseases. PDUFA VI also helps FDA to continue to advance and focus on the development and approval of medicines for rare diseases, including pediatric rare disorders.
21st Century Regulations for 21st Century Innovations
As science advances, so must the methods and tools used by the FDA to regulate drug development and review new drug applications. PDUFA VI helps encourage the use and regulatory acceptance of “21st Century” approaches to drug development, including the use of innovative clinical trial designs, biomarkers, real-world evidence, as well as other novel drug development tools.
Integration of Patient Perspective
Facilitating the inclusion of patient perspectives into drug development helps to ensure that medicines better reflect measures that are meaningful to patients. PDUFA VI strengthens the FDA’s ability to advance the science of patient input in the drug development and regulatory review processes. For example, dedicated experts will be placed into review divisions to engage with patients, patient advocates, and sponsors during drug development. The FDA will also work with patient advocates, industry, and other stakeholders on patient-reported outcomes (PROs).
Post-Approval Safety Focus
PDUFA VI will further enhance FDA’s drug safety and post-market surveillance tools and technology, including the expansion of FDA’s Sentinel Initiative.
The FDA under Commissioner Scott Gottlieb’s leadership has already begun taking steps to enhance the drug development and review processes to encourage efficiencies and competition to the benefit of patients. America’s biopharmaceutical companies are supportive of initiatives which prioritize patients and efficiencies in the drug development process. PDUFA VI helps provide much-needed resources to the agency to meaningfully advance these efforts.


